 |
Your changes have been successfully saved |

|
المركز الوطني لبلاغات الأجهزة والمنتجات الطبية
|
|
|
|
 |
|
 |
|
FAQs
National Center for Medical Device Reporting (NCMDR) is an organization managing a database of information on safety and
performance related aspects of medical devices and capable of taking appropriate action on any confirmed problems.
What are the goals of NCMDR?
Establishment of NCMDR was in order to achieve following goals to:
- Improve protection of the health and safety of patients, users and others.
- Disseminate relevant device related information which may reduce the likelihood of, or prevent repetition of adverse events,
or alleviate consequences of such repetition.
- Execute a key aspect of the SFDA's post-market activities.
- Encourage collaboration between manufacturers and health care facilities to identify and investigate adverse events
associated with medical devices and take appropriate action.
- Encourage the reporting of adverse events by medical device institutions and users, manufacturers, authorized
representatives and organizations involved in supplying medical devices to the KSA.
- Provide a database of information on the safety and performance of medical devices that is suitable for
the exchange of adverse events information with other Regulatory Authorities.
What is the important of NCMDR?
As part of its public mandate to protect patients and health professionals, Saudi Food and Drug Authority (SFDA)
maintains an online medical devices reporting system. The purpose of the system is to identify events in which:
- A patient, staff member, or visitor may have suffered harm as a result of failure or misuse of a medical device
- Medical devices fail and harm the environment
- Significant medical devices failures occur and are likely to recur and harm people or the surroundings.
What is an adverse event?
It is the report of any event that leads or might lead to undesired or unexpected issues regarding safety of patients,
users, janitors or any other person. This may include the following:
- Problems with the design of the device
- Untrained or unaware staff
- Carelessness in use
- Uninstructed modifications
- Inadequate maintenance
- Inappropriate conditions: storage, temperature and use
1) An Event has Occurred
Typical events are:
-
A malfunction or deterioration in the characteristics or performance.
A malfunction or deterioration should be understood as a failure of a device to perform in accordance with its
intended purpose when used in accordance with the manufacturer's instructions.
The intended purpose means the use for which the device is intended according to the data supplied by the manufacturer
on the labeling, in the instructions and/or in promotional materials.
-
An inadequate design or manufacture.
This would include cases where the design or manufacturing of a device is found deficient.
-
An inaccuracy in the labeling, instructions for use and/or promotional materials. Inaccuracies include omissions and deficiencies.
Omissions do not include the absence of information that should generally be known by the intended users.
-
A significant public health concern.
This can include an event that is of significant and unexpected nature such that it becomes alarming as a potential
public health hazard, e.g. human immunodeficiency virus (HIV) or Creutzfeldt-Jacob Disease (CJD).
-
Use Error
2) The Event Led to One of the Following Outcomes
-
Death of a Patient, User or Other Person.
-
Serious Injury of a Patient, User or Other Person.
Serious injury (also known as serious deterioration in state of health) is either:
- Life threatening illness or injury.
- Permanent impairment of a body function or permanent damage to a body structure.
A condition necessitating medical or surgical intervention to prevent permanent impairment of a body function or permanent damage to a body structure.
The interpretation of the term "serious" is not easy, and should be made in consultation with a medical practitioner when appropriate.
The term "permanent" means irreversible impairment or damage to a body structure or function, excluding minor impairment or damage.
Medical intervention is not in itself a serious injury. It is the reason that motivated the medical intervention that should be used to assess the report ability of an event.
-
No Death or Serious Injury Occurred but the Event Might Lead to Death or Serious Injury of a Patient, User or Other Person if the Adverse Event Recurs.
All events do not lead to a death or serious injury. The non-occurrence of such a result might have been due to circumstances or to the timely intervention of health care personnel.
The event is considered "adverse" if in the case of reoccurrence, it could lead to death or serious injury.
This applies also if the examination of the device or a deficiency in the information supplied with the device,
or any information associated with the device, indicates some factor which could lead to an event involving death or serious injury.
Include relevant information that might impact the understanding or evaluation of the adverse event AND that is not included elsewhere in this report.
3) The Manufacturer's Device is Associated with the Event
In assessing the link between the device and the event, the manufacturer should take into account:
- The opinion, based on available information, from a healthcare professional
- Information concerning previous, similar events
- Other information held by the manufacturer
This judgment may be difficult when there are multiple devices and drugs involved. In complex situations, it should be assumed that the
device was associated with the event.
A recall is an action taken to address a problem with a medical device that violates SFDA law. Recalls occur when a
medical device is defective, when it could be a risk to health, or when it is both defective and a risk to health.
A medical device recall doesn't always mean that you must stop using the product or return it to the company.
A recall sometimes means that the medical device needs to be checked, adjusted, or fixed.
Examples of the types of actions that may be considered recalls:
- Inspecting the device for problems
- Repairing the device
- Calibrating and adjusting settings on the device
- Re-labeling the device
- Destroying device
- Notifying patients of a problem
SFDA classifies medical device recalls into three categories, representing the potential risk to public health:
- Class I High risk
- Class II Medium risk
- Class III Low risk
Who should make a recall?
In most cases, a establishment (manufacturer, authorized representative, importer and distributor, or other
responsible party) recalls a medical device on its own (voluntarily). When a establishment learns that it has
a product that violates SFDA law, it
- Recalls the device(through correction or removal from service); and
- Notifies SFDA.
Legally, SFDA can require a establishment to recall a device. This could happen if a establishment refuses to recall a
device that is associated with significant health problems or death.
When an establishment recalls a medical device, it
-
Contacts directly the customers who received the product from them, and takes steps to reach others who need to be
notified (for example, by issuing press releases)
-
Supplies information to help users identify the product and take steps to minimize health consequences.
-
Takes action to prevent the problem from happening again.
How can I report an adverse event?
Device reporting can be entered by healthcare providers, manufacturers, authorized representative, distributors and public.
To enter a new device report by either healthcare providers or manufacturers, authorized representative, distributors and
public first you must click on the "Click here to Report" at home page to transfer you to new page. If you are healthcare
provider go to the first paragraph to report medical device adverse event and also the Manufacturers, authorized representatives,
importers and distributors are not required to log in prior to submitting Adverse Events Reports. However, logging in prior to
entering a Report ensures that your identifying information is automatically entered. (e.g., e-mail address, name, organization).
But if you logged in, manufacturers, authorized representatives and distributors can click on the Device report tab on the top menu bar.
The device report entry page is divided into two sections. The first section contains your personal information which is
pre-populated with information in your profile. You can make any changes you may wish. All fields marked with a red asterisks (*) are required.
Device Information
The Device Information section (Figure 1) provides NCMDR information about the device that is involved in the adverse event.
All fields should be entered as completely as possible. All fields marked with a red asterisks (*) are required

Figure 1: Device Information
|
Field Name
|
Required
|
Description
|
|
Type(s) of device(s) involved
|
Required
|
The type of devices that were involved in the event.
|
|
Manufacture
|
Required
|
The manufacture of the device.
|
|
Local Supplier/Distributor
|
Required
|
The local supplier or distributor of the device
|
|
Serial/Lot No
|
Required
|
The serial number or lot number of the device
|
|
Expiration/Used Before Date
|
Optional
|
If the device is a single-use device, enter the expiration or Use Before Date
|
|
How long in use
|
Optional
|
Enter how long the device was in use.
Ex.: 4 years 3 months
|
|
Condition
|
Optional
|
The condition of the device at the time of the incident. Ex.: Brand new
|
|
Date Adverse Event occurred
|
Required
|
The date the Adverse Event occurred (this should be enter in Common Era year)
|
|
Date last inspected or serviced
|
Optional
|
Date the device was last inspected or serviced
|
|
Where other devices involved?
|
Optional
|
If other devices were involved in the incident, check Yes and enter the
devices in the box below. If there
were no other devices involved, check No.
|
|
If yes, please describe
|
Optional
|
If there were other devices involved in the Adverse Event, enter the devices
here.
|
|
Is the device available for inspection?
|
Optional
|
Check Yes if SFDA is able to inspect the device. Check No, if the device is
unavailable for inspection.
|
|
If a single-use device was involved...
|
Optional
|
If the device is a single-use device, indicate if the device was reprocessed.
|
|
Are other units of the same model similarly affected?
|
Optional
|
Indicate if you are experiencing similar Adverse Event with other units of the
same model.
|
Adverse Event Description
Please use the text box (Figure 2) to describe the hazard or adverse event in detail. Include how it was discovered,
any action you took, and the response of any manufacturers , authorized representative and distributors.
Please also mail or fax any related correspondence when possible. Sketches, photographs, or copies of portions of
operating manuals are often helpful in describing the Adverse Event, especially if the affected device is not available
for examination at SFDA. Retain all disposable accessories involved in an incident. Please do not send any devices to
SFDA until requested. All fields marked with a red asterisks (*) are required.

Figure 2: Adverse Event Description
|
Field
Name
|
Required
|
Description
|
|
Outcome
of the Adverse Event
|
Optional
|
If the
Adverse Event resulted in death, injury, or near miss, check the appropriate
button. Otherwise, please click
Other and briefly describe the outcome in the box immediately below.
|
|
Adverse
Event Details
|
Required
|
Please
enter details of the Adverse Event.
The more relevant details you provide, the quicker SFDA can assess the Adverse
Event.
|
After all three sections of the adverse event report have been completed, The item will enter into the system for
investigation as well as an E-mail will be automatically sent to the reporter with the Confirmation Code and a link
to access the adverse event report. The Confirmation Code allows users to access previously entered Adverse events
report (Figure 3). When accessing previously entered Adverse events reports, additional information can be appended
to the original description. These information do not alter information entered initially. NCMDR staff may modify
the original text to conform with industry standard nomenclature.
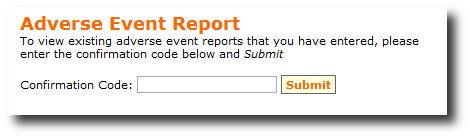
Figure 3: viewing Pre-entered Problem Report
How can an establishment makes a recall?
All medical-devices manufacturers , authorized representative, importer and distributors must first register
(Figure 4) in order to enter Devices Recalls. After registering, the SFDA Administrator reviews the information
and activates the account. Then, a manufacturer , authorized representative, importer and distributors is able to
log into the account.

Figure 4: Registration Form
All manufacturers , authorized representative,importer and distributors must log in to view existing Devices Recalls
or enter new Devices Recalls (Figure 5). Manufacturers , authorized representative and distributors are not required
to log in prior to submitting Problem Reports. However, logging in prior to entering a Report ensures that your
identifying information is automatically entered. (e.g., e-mail address, name, organization)
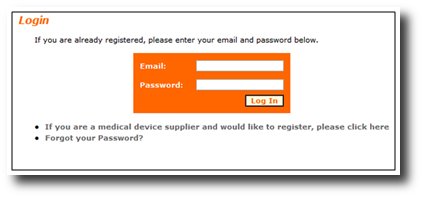
Figure 5: Logging In
Is it safe to report (Confidentiality)?
SFDA will share all submitted information whether recalls or reports with ECRI and other global authorities,
and SFDA will ensure to hide the identity of the person who submitted the information and will not share personal
data with any other parties. All personal data will be used within SFDA only.
What is a medical device?
Medical device: means any instrument, apparatus, implement, machine, appliance, implant, in vitro reagent or
calibrator, software, material or other similar or related article:
Intended by the manufacturer to be used, alone or in combination, for human beings for one or
more of the specific purpose(s) of:
- diagnosis, prevention, monitoring, treatment or alleviation of disease,
- diagnosis, monitoring, treatment, alleviation of or compensation for an injury or handicap,
- investigation, replacement, modification, or support of the anatomy or of a physiological process,
- supporting or sustaining life,
- control of conception,
- disinfection of medical devices,
- providing information for medical or diagnostic purposes by means of in vitro examination of specimens derived
from the human body; and which does not achieve its primary intended action in or on the human body by pharmacological,
immunological or metabolic means, but which may be assisted in its intended function by such means.
What is the in vitro diagnostic device(IVDD)?
In-vitro medical device: means a medical device, whether used alone or in combination, intended by the manufacturer
for the in-vitro examination of specimens derived from the human body solely or principally to provide information for
diagnostic, monitoring or compatibility purposes. This includes reagents, calibrators, control materials, specimen
receptacles, software and related instruments or apparatus or other articles.
What is medical devices accessories?
Accessory: means a product intended specifically by its manufacturer to be used together with a medical device to
enable that medical device to achieve its intended purpose.
|
|
 |
|
 |
|
|